Extensive simulations assess the performance of genome-wide association mapping in various Saccharomyces cerevisiae subpopulations.
Jackson Peter, Anne Friedrich, Gianni Liti, Joseph Schacherer.
Philosophical Proceedings of the Royal Society B. 2022. doi:10.1098/rstb.2020.0514
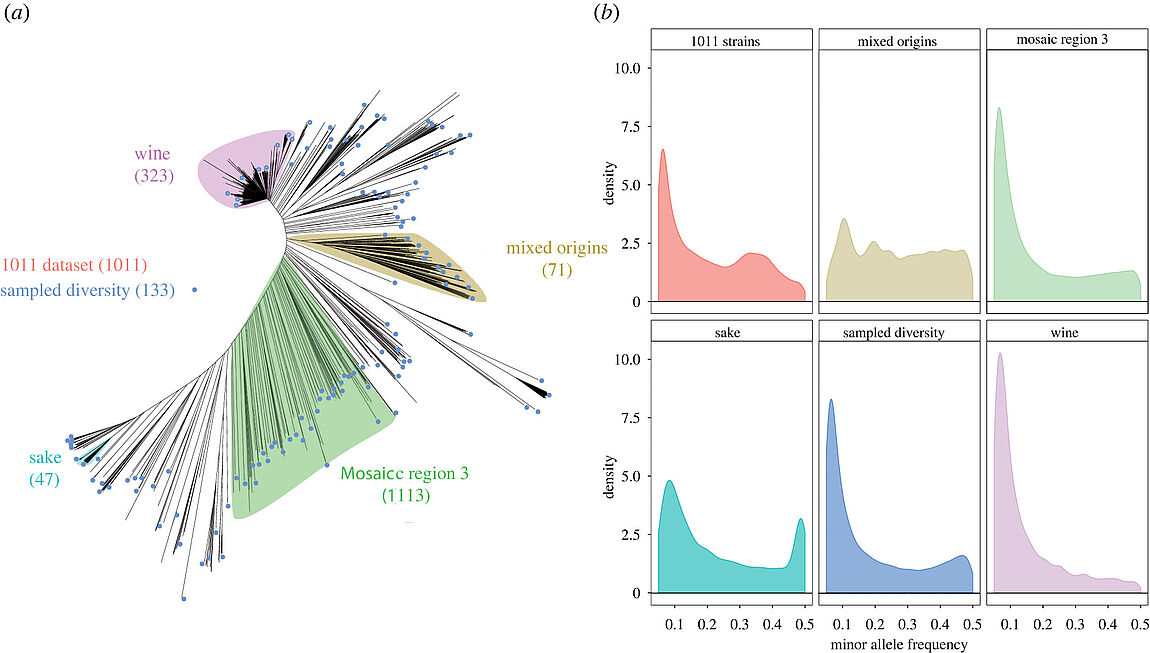
With the advent of high throughput sequencing technologies, genome-wide association studies (GWAS) have become a powerful paradigm for dissecting the genetic origins of the observed phenotypic variation. We recently completely sequenced the genome of 1011 Saccharomyces cerevisiae isolates, laying a strong foundation for GWAS. To assess the feasibility and the limits of this approach, we performed extensive simulations using five selected subpopulations as well as the total set of 1011 genomes. We measured the ability to detect the causal genetic variants involved in Mendelian and more complex traits using a linear mixed model approach. The results showed that population structure is well accounted for and is not the main problem when the sample size is high enough. While the genetic determinant of a Mendelian trait is easily mapped in all studied subpopulations, discrepancies are seen between datasets when performing GWAS on a complex trait in terms of detection, false positive and false negative rate. Finally, we performed GWAS on the different defined subpopulations using a real quantitative trait (resistance to copper sulfate) and showed the feasibility of this approach. The performance of each dataset depends simultaneously on several factors such as sample size, relatedness and population evolutionary history.